Highly accurate protein structure prediction with AlphaFold
Jumper, Evans, Pritzel, Hassabis et al. — Nature 2021 (DeepMind)
蛋白质折叠问题——给定氨基酸序列,预测其三维结构——困扰了生物学界 50 年。2020 年,DeepMind 的 AlphaFold2 在 CASP14 竞赛中将预测误差控制在原子精度(小于 1.5 埃),远超所有对手(普遍 3 埃左右),被誉为 AI 在科学领域最重大的突破之一。Hassabis 和 Jumper 因此获得 2024 年诺贝尔化学奖。
研究动机
蛋白质的三维结构决定了其生物功能,但实验测定结构(X 射线晶体学、冷冻电镜)耗时数月甚至数年。计算预测方法长期受限于两个核心问题:
- 搜索空间爆炸:一个 200 个氨基酸的蛋白质,其构象空间是天文数字。传统物理模拟(分子动力学)无法在合理时间内搜索
- 进化信息利用不足:共进化分析虽能推断残基空间邻近关系,但此前的方法未能端到端地将其整合进结构预测
AlphaFold2 将蛋白质折叠重新定义为进化信息 + 几何空间上的图推理问题,从头设计了一套能同时理解"进化语言"和"三维几何语言"的端到端神经网络架构。
整体架构

AlphaFold2 的架构分为三大模块:
| 模块 | 功能 | 规模 |
|---|---|---|
| 特征提取 | 搜集进化信息(MSA)+ 结构模板 | 数据库搜索 |
| Evoformer 编码器 | 提取氨基酸之间的深层关系 | 48 个 Transformer Block |
| 结构模块解码器 | 预测蛋白质 3D 结构 | 8 个共享权重 Block |
此外,模型采用回收机制(Recycling):将上一轮推理的输出作为下一轮的输入,循环 3 次让预测逐轮精化,类似 RNN 的思想但仅在第一轮更新梯度。
特征提取
MSA:挖掘进化信息
使用 jackhmmer 和 HHblits 搜索多个序列数据库(UniRef90、MGnify、BFD 等),找到数千条进化上相关的同源序列,构建多序列比对(MSA)——每一行是一条蛋白质序列,每一列是序列中的一个位置。
MSA 的核心价值:
- 保守性分析:某一列绝大多数氨基酸相同 → 该位置在进化中高度保守,对结构或功能至关重要
- 共进化分析:两列的氨基酸变化具有强相关性 → 这两个位置在空间上可能邻近
生成的 MSA 表示是一个 三维张量(序列数量、序列长度、特征维度)。
配对表示:几何蓝图
模型需要一个描述"任意两个氨基酸之间关系"的特征矩阵——配对表示 。可以想象成一个 的矩阵,每个位置 存储一个高维向量,描述第 和第 个氨基酸的关系。
信息来源:MSA 共进化信号 + 结构模板的距离矩阵 + 位置编码(告诉模型 和 在序列中的相对距离 )。
Evoformer 编码器
Evoformer 是 AlphaFold2 的核心,与标准 Transformer 的关键区别在于:输入是 MSA 表示和配对表示两个二维矩阵,而非一维序列。48 个 Block 逐步精炼两个表示之间的关系。
行注意力(Row-wise Gated Self-Attention with Pair Bias)
对 MSA 的每一行执行自注意力,同时将配对表示作为注意力偏置注入:
其中 从配对表示线性投影得到,提供"残基 和 的空间关系提示"。输出经过 Sigmoid 门控 逐元素调节:
列注意力与 Transition
列注意力沿序列维度(不同同源序列之间)进行信息混合,捕捉不同物种在同一位置上的变异模式——共进化信号的直接来源。列注意力不加配对偏置。
Transition 是标准前馈网络(128 → 512 → ReLU → 128),负责信息的深度加工和提炼。
Outer Product Mean(外积均值)
将一维序列特征"升维"为二维配对关系特征的专用模块:
- Mean:沿 维度平均,将数千条同源序列压缩为一条"共识序列"
- Outer Product:对任意位置对 ,计算特征向量的外积 ,生成 的矩阵
外积矩阵编码了"哪些氨基酸倾向于协同变化" → "哪些氨基酸在空间上可能相互接触"。
三角自注意力(Triangle Self-Attention)
AlphaFold2 独有的几何推理模块。蛋白质中三个残基 的距离满足三角不等式:。三角注意力将边(而非节点)作为处理对象:
- 选择起始节点 ,将所有边 组成序列
- Query 来自边 ,Key/Value 来自边
- 更新后的信息反映"从 的视角看, 和 的三角一致性"
另有方向相反的 Around Ending Node 模块,确保信息双向流动。
编码器输出
| 精炼后的配对表示 | 单序列表示 | |
|---|---|---|
| 回答的问题 | 氨基酸 和 的关系是怎样的? | 氨基酸 自身的状态和环境? |
| 数据结构 | 的关系地图 | 长度为 的状态列表 |
| 核心信息 | 距离、方向等几何关系 | 局部结构环境(螺旋/折叠) |
结构模块与 IPA 解码器
结构模块由 8 个共享参数的 Block 组成(类似 RNN),将编码器输出的抽象"几何蓝图"逐步转化为精确的三维坐标。
解码流程
- 初始化:所有氨基酸在三维空间原点 ,一团重叠的"原子云"
- IPA 定位:每个氨基酸问三个问题——"我是谁?"(查单序列表示)"我的邻居在哪?"(查配对表示)"我现在在哪?"(查当前 3D 坐标)
- 迭代精修:重复 8 次,每次让结构更精确、能量更低
- 最终输出:每个原子的 坐标 + pLDDT 置信度分数(0-100)
Invariant Point Attention(IPA)
IPA 是结构模块的核心创新:
- Point(点):处理三维空间中的原子坐标
- Invariant(不变):计算结果对全局旋转和平移保持不变——将蛋白质平移 100 埃或旋转 90 度,IPA 的输出完全相同
- Attention:注意力机制的特殊改造版本
实现不变性的关键:为每个残基建立独立的局部坐标系,所有计算在局部坐标系中完成:
- 为点 创建局部坐标系(基于主链原子位置)
- 将其他点 的信息转换到 的局部坐标系
- 在局部坐标系中完成注意力计算
- 将局部更新指令翻译回全局坐标系
注意力权重混合三方面信息:
其中 将局部坐标点转换到全局坐标系。三个分量分别编码:单序列表示的相似度("理论上的关系")、配对表示的几何蓝图、当前 3D 空间中的实际距离。
总结
AlphaFold2 的根本创新在于将蛋白质折叠问题重新定义为进化信息和几何空间上的图推理问题。Evoformer 通过行/列注意力、外积均值和三角注意力同时处理 MSA 和配对表示;IPA 通过局部坐标系保证 SE(3) 不变性,在 8 轮迭代中将抽象蓝图"打印"成精确的三维结构。
代码实战
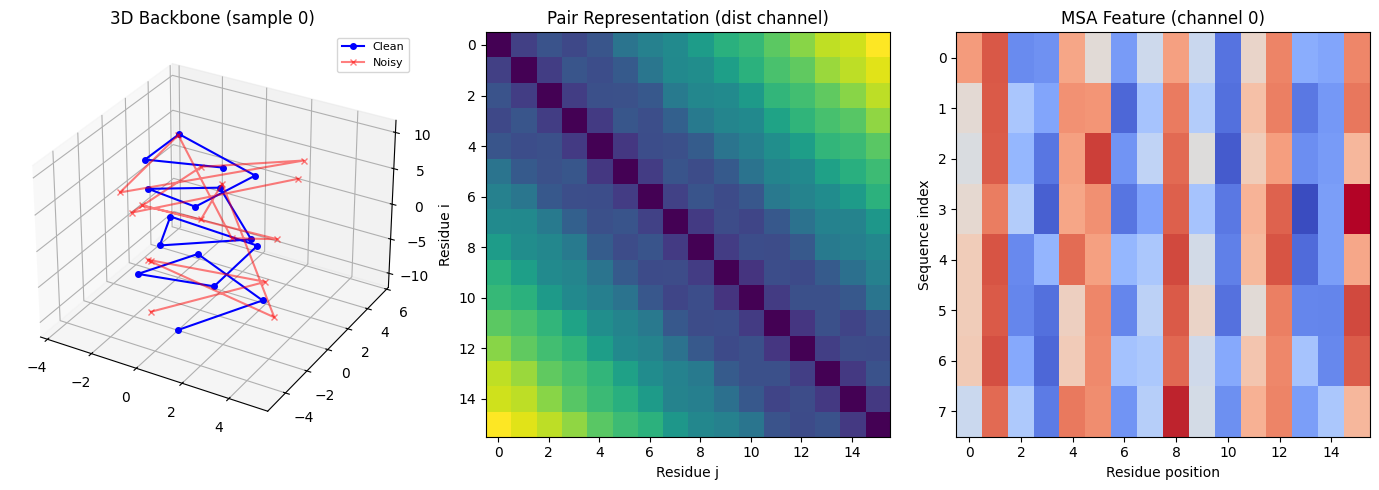
完整代码包含从零手写 Evoformer + IPA 和使用 invariant-point-attention 库的简洁实现,以蛋白质骨架坐标去噪任务验证核心架构:
![]()
合成数据与任务
用参数化的 -螺旋和 -折叠几何构建合成蛋白质骨架,加噪后让模型恢复原始坐标——模拟 AlphaFold2 结构模块从"原子云"迭代精修到精确结构的核心行为。
行注意力 + 配对偏置
Evoformer 的核心模块——将配对表示 作为注意力偏置注入,让几何信息引导序列特征更新:
class RowAttentionWithPairBias(nn.Module):
def forward(self, msa, pair):
# QKV 投影
q, k, v = self.to_qkv(self.norm(msa)).unbind(dim=3)
# 注意力分数 + 配对偏置
attn = (q @ k.T) * self.scale
bias = self.to_bias(pair) # 从配对表示提取几何提示
attn = (attn + bias).softmax(dim=-1)
# 门控聚合
gate = sigmoid(self.to_gate(msa))
return msa + gate * (attn @ v)IPA 手写实现
IPA 的注意力权重混合标量通道、配对偏置和点通道(3D 空间距离):
# 标量通道:单序列表示的相似度
scalar_attn = (q @ k.T) * self.scale
# 配对偏置:几何蓝图信息
pair_bias = self.to_bias(pair)
# 点通道:T_i(p) = R_i @ p + t_i,在全局坐标系中计算距离
q_pts_global = R_i @ q_pts + t_i
k_pts_global = R_j @ k_pts + t_j
pt_attn = -gamma * ||q_pts_global - k_pts_global||^2
# 合并三个通道
attn = softmax(scalar_attn + pair_bias + pt_attn)训练结果
两种实现在坐标去噪任务上均收敛至亚埃级精度:
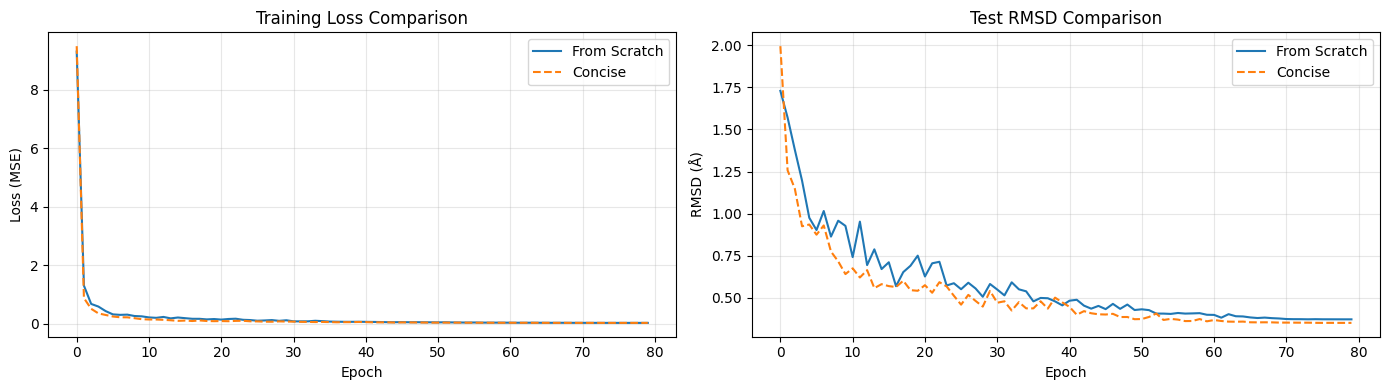

参考文献
- Jumper, J., Evans, R., Pritzel, A., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596, 583-589.
- 李沐. AlphaFold2 论文精读. Bilibili.
- lucidrains. invariant-point-attention. GitHub.